
Nature Methods:Chip-Tip技术——突破单细胞蛋白质组学研究瓶颈的革命性进展
来源:生物探索 2025-01-23 09:43
这项研究不仅推动了单细胞蛋白质组学的技术发展,也为生物医学研究中细胞功能和疾病机制的深入理解奠定了基础。
随着生命科学领域的快速发展,研究人员已经不再仅仅依赖传统的群体样本分析,而是开始深入探索单个细胞的复杂性。单细胞蛋白质组学(Single-Cell Proteomics, SCP)作为一项前沿技术,正在为我们提供前所未有的机会,直接揭示单个细胞内的蛋白质组成和功能状态。这一技术不仅能够揭示细胞在不同生物学过程中的动态变化,还能帮助我们更深入地理解疾病的机制,特别是在癌症、免疫和神经退行性疾病等领域的潜在应用。
然而,单细胞蛋白质组学的广泛应用面临着许多技术挑战,其中最为突出的是分析的灵敏度、样本处理的复杂性以及数据分析的高难度。为了突破这些障碍,研究人员们不断努力优化现有的技术手段,提升单细胞分析的准确性和高通量处理能力。
1月16日Nature Methods的研究报道“Enhanced sensitivity and scalability with a Chip-Tip workflow enables deep single-cell proteomics”,研究团队提出了一种创新的单细胞蛋白质组学工作流程——“Chip-Tip”方法,显著提高了蛋白质的识别灵敏度和分析的深度。这项技术利用了新型的ProteoCHIP EVO 96芯片进行单细胞样本的高效制备,通过直接转移和清洗步骤,结合Evosep One液相色谱系统和Orbitrap Astral质谱仪,极大提升了样本分析的通量和精确度。研究人员在单个HeLa细胞中成功识别了超过5000种蛋白质,并且能够直接检测到细胞中的翻译后修饰(PTMs),如磷酸化和糖基化,无需进行额外的修饰富集步骤,这一突破为研究细胞的功能提供了更加全面的视角。
此外,研究团队还扩展了该方法的应用,分析了人源诱导多能干细胞(hiPSCs)的分化过程,并识别出了多个细胞谱系特异性的标志物。这项研究不仅推动了单细胞蛋白质组学的技术发展,也为生物医学研究中细胞功能和疾病机制的深入理解奠定了基础。

单细胞蛋白质组学:揭开细胞奥秘的钥匙
单细胞蛋白质组学(Single-Cell Proteomics, SCP)是一项能够深入探索单个细胞内蛋白质组的前沿技术。与传统的群体细胞研究不同,单细胞蛋白质组学为我们提供了一个更精细的视角,让我们能够理解每个细胞在复杂生物体中的独特功能和行为。这项技术不仅为基础生物学研究提供了新的思路,还在癌症、免疫学和神经科学等领域的临床研究中展现出巨大的潜力。通过揭示不同细胞在相同环境下的差异,我们能够更好地理解细胞分化、疾病发展以及药物反应等生命过程。
尽管单细胞蛋白质组学为生命科学研究带来了革命性进展,但其实施过程依然面临着诸多挑战。首先,单细胞中的蛋白质数量极为稀少,这使得传统的蛋白质分析方法往往难以检测到所有的目标蛋白质。在早期的单细胞蛋白质组学研究中,研究人员通常只能识别出几百个蛋白质,且这些蛋白质往往集中在丰度较高的蛋白质上,而低丰度的蛋白质则常常被忽视。这种数据的偏差限制了我们对细胞功能全面了解的能力。
其次,单细胞样本的制备过程非常复杂。细胞裂解、蛋白质提取和消化等步骤都容易引入样本损失,影响最终的分析结果。传统方法中,细胞破裂后的蛋白质可能因表面吸附而丢失,进一步限制了我们对细胞内蛋白质的深度探测。此外,单细胞蛋白质组学还面临着高通量和高灵敏度之间的平衡问题。单个细胞的样本量极其微小,如何在保证高灵敏度的同时,还能进行高通量的分析,成为了科研人员的一大难题。
在数据分析方面,如何从海量的质谱数据中提取出有意义的信息也是一个亟待解决的问题。由于单细胞样本中的蛋白质数量较少,且与多细胞样本相比背景噪音较大,传统的数据处理算法往往无法有效地识别低丰度蛋白质和翻译后修饰(PTMs)。这些技术瓶颈使得单细胞蛋白质组学的应用受到了限制。
“Chip-Tip”方法:突破单细胞分析的技术瓶颈
“Chip-Tip”方法是一种创新的单细胞蛋白质组学分析技术,旨在突破单细胞研究中常见的灵敏度、通量和样本损失等技术瓶颈。该方法通过一系列精细优化的步骤,将细胞样本制备、蛋白质提取与质谱分析有机结合,极大提升了单细胞分析的深度和广度。
这一方法的核心在于ProteoCHIP EVO 96芯片,它能够在微纳体积下高效处理单个细胞。首先,细胞被精确分配到芯片上的小孔内,利用微流控技术进行细胞裂解和蛋白质提取。与传统的处理方法不同,这一过程减少了表面吸附和样本损失,从而保证了最大化的蛋白质提取率和分析深度。经过这一预处理,样本会直接转移到Evosep One液相色谱系统(LC)进行分离,再结合Orbitrap Astral质谱仪(MS)进行高灵敏度的分析。
通过这种无缝衔接的流程,研究人员能够在单细胞水平上获取高质量的蛋白质组数据,识别出更多的蛋白质和翻译后修饰(PTMs)。这一技术的突破不仅增强了分析的灵敏度,还实现了更高的通量,使得研究者能够在一天内处理多达120个单细胞样本,显著提升了数据采集效率。
技术创新:从样本准备到数据分析的革新
“Chip-Tip”方法在技术上具有多个创新亮点,特别是在样本准备和数据分析阶段。首先,ProteoCHIP EVO 96芯片的设计使得单细胞蛋白质样本的制备更加高效和无损。芯片的每个孔能够精确处理1-96个细胞,这意味着在同一实验中,研究人员可以同时处理大量的单细胞样本,减少了实验周期和人工干预的误差。此外,芯片采用纳升级别的微流控技术,能够最大限度地减少样本损失,避免了传统方法中常见的样本吸附和分析误差。
随后,样本会被转移至Evosep One液相色谱系统进行高效分离。在这一过程中,Evosep One利用了Whisper flow技术,通过精确控制流速和温度,确保了分离过程的高效性和准确性。与传统的液相色谱系统相比,Evosep One系统在处理单细胞样本时显示出更高的灵敏度和分辨率,从而可以识别更多的低丰度蛋白质。
最后,质谱分析阶段采用的是Orbitrap Astral质谱仪,它结合了最新的nDIA技术,通过优化的扫描模式和高精度的离子捕获方法,使得在单细胞蛋白质组学分析中取得了前所未有的深度和灵敏度。研究表明,使用这一系统,研究人员能够在单个HeLa细胞中识别出超过5000种蛋白质,且这些蛋白质的序列覆盖率和丰度范围均表现出极高的一致性和准确性。
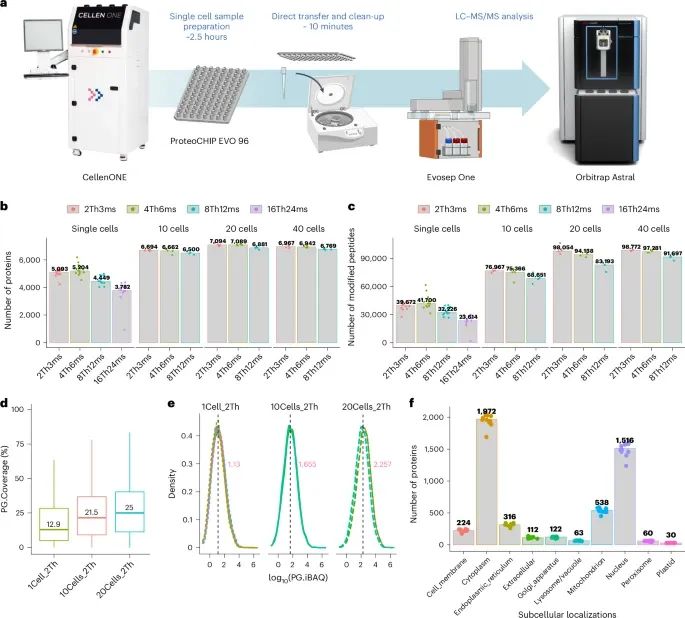
Chip-Tip SCP工作流程与结果(Credit: Nature Methods)
a. Chip-Tip工作流程示意图:展示了Chip-Tip方法的总体流程,包括单细胞样本的处理、ProteoCHIP EVO 96芯片用于细胞裂解和蛋白质提取、样本通过Evosep One液相色谱系统分离后进行Orbitrap Astral质谱分析等步骤。通过这一流程,研究人员能够高效地识别和量化单细胞中的蛋白质。
b. 不同细胞数量下识别的蛋白质数量:展示了在单个、10个、20个和40个HeLa细胞样本中,使用不同的nDIA(narrow-window data-independent acquisition)设置时所识别的蛋白质数量。数据显示,随着细胞数量的增加,识别的蛋白质数量呈显著上升趋势,单细胞样本中识别到约5204个蛋白质,而20个细胞样本中识别到超过7000个蛋白质。
c. 不同细胞数量下识别的肽段数量:展示了在单细胞、10个、20个和40个细胞样本中,使用nDIA设置时识别的肽段数量。数据显示,细胞数量的增加同样导致肽段数量的增加,单细胞样本中识别到约41,700个肽段,20个细胞样本中则增加到98,054个肽段。
d. 蛋白质序列覆盖率:展示了单细胞、10个细胞和20个细胞样本中的蛋白质序列覆盖率。数据表明,随着样本细胞数的增加,蛋白质的序列覆盖率也有所提升,20个细胞样本的序列覆盖率达到了25%。
e. iBAQ值分布:iBAQ(intensity-based absolute quantification)值反映了蛋白质定量范围及一致性,该部分展示了不同细胞数量下蛋白质的定量范围。结果显示,不同细胞数量样本中的蛋白质定量值在iBAQ范围内表现出较好的一致性,验证了方法的可靠性。
f. 蛋白质在细胞不同部位的定位:展示了所识别蛋白质在细胞内各个亚细胞区室的定位分布。研究发现,超过200种蛋白质定位于细胞膜,显示了该方法在蛋白质组学分析中的全面性和准确性。
高灵敏度与高通量:如何实现蛋白质的深度分析
蛋白质识别:从单个细胞到20个细胞的蛋白质量化
在单细胞蛋白质组学中,蛋白质的精确识别一直是一个难题,尤其是在蛋白质丰度较低的情况下。传统方法常常无法有效捕捉到这些低丰度蛋白质,导致蛋白质组覆盖度较低。然而,通过“Chip-Tip”方法,研究人员突破了这一瓶颈,实现了从单个细胞到20个细胞的高效蛋白质识别。
研究数据显示,使用这一技术,单个HeLa细胞中能够识别出超过5000种蛋白质,而在20个细胞的样本中,蛋白质识别数量超过了7000种。这一突破显示,随着细胞数目的增加,蛋白质的识别数量呈现出明显的增长,且随着样本量的扩大,蛋白质的多样性得到了显著提升。
此外,通过使用nDIA(narrow-window data-independent acquisition)质谱扫描模式,研究人员能够在每个细胞中实现对蛋白质的深度挖掘,确保了即使是低丰度的蛋白质也能被准确捕捉。这不仅提高了蛋白质组的识别数量,还为进一步的功能分析和疾病机制研究提供了更多数据支持。
高深度蛋白质组学:如何在单细胞分析中实现前所未有的灵敏度
“Chip-Tip”方法的核心优势之一便是其在深度分析方面的卓越表现。研究表明,通过这一方法,单个细胞的蛋白质组覆盖度达到了前所未有的深度。具体来说,在单细胞样本中,研究人员能够获得超过41,000个肽段的标定数据,这使得对每个蛋白质的分析更加全面和精准。尤其是在低丰度蛋白质的检测方面,这一技术展现了无与伦比的灵敏度。
例如,在磷酸化修饰分析中,研究人员在单个细胞中直接识别出了超过120个磷酸化位点,涵盖了包括丝氨酸(Ser)、苏氨酸(Thr)和酪氨酸(Tyr)在内的多个关键磷酸化位点。这一发现表明,单细胞蛋白质组学不仅能提供蛋白质识别的数据,还能够深入探测细胞内的翻译后修饰(PTMs),进而揭示细胞在不同生物过程中的功能状态。这种深度的分析能力在传统方法中是难以实现的,尤其是在无需额外富集步骤的情况下,仍能准确捕捉到翻译后修饰的信息。
此外,随着样本量的增加,蛋白质组的覆盖率也有了显著提升。在20个细胞样本中,研究人员能够识别出高达98,000个肽段,极大提高了数据的可靠性和深度。这一高深度的分析为研究人员提供了更加丰富的细胞内信息,帮助他们更好地理解细胞的生物学功能以及疾病的分子机制。
直接揭示翻译后修饰(PTMs):一个革命性的突破
磷酸化与糖基化:无需额外富集步骤的检测
翻译后修饰(Post-translational Modifications, PTMs)是蛋白质功能调控的关键机制,涉及磷酸化、糖基化、乙酰化等多种化学修饰。磷酸化和糖基化是细胞内两种最为常见且功能至关重要的翻译后修饰。磷酸化调控着蛋白质的活性、稳定性、位置以及与其他分子的相互作用,而糖基化则在细胞信号传导、免疫反应和细胞通讯中发挥着重要作用。
传统的PTM分析方法通常需要对样本进行特定的富集步骤,以便检测这些低丰度的修饰位点。例如,磷酸化通常需要通过磷酸化抗体进行富集,糖基化则通过特定的糖基化抗体或化学标签进行富集。然而,这些步骤不仅增加了实验的复杂性,还可能导致某些低丰度修饰的丢失。
“Chip-Tip”方法的创新之处在于,它能够直接在单细胞样本中进行PTMs分析,无需任何额外的富集步骤。通过这种方法,研究人员能够更高效地识别细胞中的磷酸化和糖基化位点,且在无需繁琐富集的情况下,依然能够获得高质量的数据。这为PTM研究开辟了新的方向,使得我们能够更全面、准确地了解翻译后修饰对细胞功能的调控。
如何在单细胞中直接识别翻译后修饰
“Chip-Tip”方法的引入,使得在单细胞层面上对磷酸化和糖基化的识别变得更加直接和精确。研究人员在单个细胞样本中,利用这一技术成功识别了多个关键的磷酸化位点,包括丝氨酸(Ser)、苏氨酸(Thr)和酪氨酸(Tyr)的磷酸化修饰。通过无富集的直接分析,研究人员在单细胞中识别到了约120个磷酸化位点,其中包括来自不同激酶家族的蛋白质,如CDK1(细胞周期蛋白依赖性激酶1)和MAPK1(丝裂原活化蛋白激酶1)等。
特别地,在糖基化修饰的识别上,研究人员在单细胞样本中直接检测到多种糖基化相关的糖基转移酶和糖基化位点。这一发现表明,“Chip-Tip”方法能够在单细胞分析中同时揭示磷酸化和糖基化等多种翻译后修饰,并且其覆盖面广泛,深度足够,使得细胞内的复杂调控机制得以全貌呈现。
以磷酸化为例,研究团队通过提取质谱数据中的特定离子,准确识别了位于不同蛋白质中的磷酸化位点,极大提升了磷酸化分析的深度和精确度。糖基化修饰的分析同样展现出巨大的优势,研究人员通过扫描特定的醣基离子(如HexNAc、NeuAc等)成功识别出细胞内多种糖基化类型,揭示了细胞内复杂的糖基化网络。
这一研究成果展示了“Chip-Tip”方法的强大能力,不仅使得PTMs分析变得更加高效和精准,也为我们深入了解细胞内的调控机制提供了新的视角。
从基础研究到临床应用:单细胞蛋白质组学的广泛潜力
单细胞分析在癌症和疾病研究中的应用
单细胞蛋白质组学(SCP)的引入为癌症和多种疾病的研究提供了全新的视角。癌症细胞的异质性使得传统的群体细胞研究方法难以全面揭示癌症发展和治疗的复杂机制。而通过单细胞分析,研究人员能够揭示不同癌细胞之间的功能差异,了解癌症在不同阶段的分子特征,进而为精准医疗的制定提供关键的数据支持。
在该研究中,研究人员通过“Chip-Tip”方法分析了癌细胞在5-氟尿嘧啶(5-FU)治疗中的反应。在处理后的肿瘤球样本中,研究人员发现,5-FU的作用不仅显著影响了细胞的结构完整性,还改变了肿瘤细胞中的多个关键蛋白质的表达。尤其是在细胞周期调控、核糖体合成及嘧啶代谢等重要生物过程中的变化,揭示了5-FU通过改变这些机制来抑制肿瘤生长。此外,研究团队还通过数据挖掘发现了肿瘤细胞对治疗的不同反应,这些发现为未来癌症治疗的靶向药物设计提供了重要依据。
单细胞蛋白质组学的应用还扩展到了其他疾病的研究中,尤其是在神经退行性疾病和免疫疾病的研究中。通过对单个免疫细胞或神经元的蛋白质分析,研究人员能够深入了解免疫反应或神经退行性病变的分子机制,为新的治疗策略的开发提供了关键的分子靶标。
从干细胞分化到癌细胞反应
除了癌症和疾病研究外,单细胞蛋白质组学还在干细胞研究中展现了巨大的应用潜力。在该研究中,研究人员采用了“Chip-Tip”方法分析了人源诱导多能干细胞(hiPSCs)在不同分化状态下的蛋白质组特征。研究人员不仅在hiPSCs中成功识别了超过4700种蛋白质,还在分化过程中成功检测到多个谱系特异性标志物,如GATA4(内胚层)、HAND1(中胚层)和MAP2(外胚层),这些蛋白质的变化为研究细胞分化提供了新的分子依据。
特别是在EBs细胞的分析中,研究人员发现,不同分化阶段的细胞展现出截然不同的蛋白质表达特征,且这种差异与特定基因的激活及细胞功能的改变密切相关。这些发现不仅加深了我们对干细胞分化的理解,还为干细胞相关疾病的治疗提供了新的研究方向。
此外,研究人员还通过分析癌细胞与干细胞的对比,展示了“Chip-Tip”方法在癌细胞反应分析中的强大能力。例如,在研究细胞暴露于抗癌药物的反应时,研究人员能够在单细胞水平上揭示药物作用后的蛋白质变化,这为精准药物筛选和个性化治疗方案的制定提供了重要参考。
未来:技术发展的方向与挑战
技术整合:单细胞蛋白质组学与其他“组学”的结合
随着单细胞技术的不断进步,单细胞蛋白质组学(SCP)与其他组学技术的结合正在推动生物学研究进入一个新的时代。单细胞RNA组学、单细胞基因组学和单细胞表观组学的整合,使得我们可以从多个维度全面解析单个细胞的功能状态。通过将这些技术结合,研究人员能够不仅仅了解细胞的基因表达,还能深入到蛋白质的功能调控和翻译后修饰等层面,从而获得更全面的细胞功能图谱。
例如,结合单细胞RNA测序和蛋白质组学,可以对转录和翻译层次进行对比分析,揭示mRNA水平和蛋白质水平之间的关联,进一步理解基因表达的复杂性和翻译后修饰的调控作用。此外,单细胞基因组学的结合使得研究人员能够探索单细胞的遗传变异及其如何影响蛋白质功能,尤其是在癌症研究中,基因突变与蛋白质变化之间的关系对于肿瘤异质性和抗药性机制的理解具有重要意义。
在未来,通过多组学技术的结合,研究人员将能够更精确地描绘细胞内外的复杂相互作用,推动精准医学和个性化治疗的发展。这种整合不仅提高了单细胞分析的深度,也为探索复杂生物现象提供了强有力的工具。
面临的挑战与未来的方向
尽管单细胞蛋白质组学在技术和应用上取得了显著进展,但它仍面临着许多挑战。首先,当前技术在样本处理和数据分析方面仍有一定的局限性。单细胞样本的处理和蛋白质提取过程需要极高的精度,且由于细胞本身的微小尺寸和低丰度蛋白质的存在,任何微小的误差都可能导致数据的丢失或偏差。尽管“Chip-Tip”方法通过精确的芯片技术减少了样本损失,但如何进一步优化样本制备流程、减少背景噪声和提高分析的准确性,仍然是未来研究的重点。
其次,数据分析是另一个亟待突破的领域。单细胞蛋白质组学的数据量巨大,如何从海量的质谱数据中提取出高质量的信息,尤其是在低丰度蛋白质和翻译后修饰的检测方面,需要更加先进的算法和计算工具。随着数据分析技术的不断提升,机器学习和人工智能的引入可能会成为解决这一问题的关键,通过智能化的算法提高数据处理的效率和准确性。
此外,单细胞分析的通量问题仍然是制约其广泛应用的瓶颈之一。尽管当前技术已能处理大量样本,但如何进一步提高样本处理的速度和规模,使得单细胞分析能够在临床诊断和大规模筛查中得到应用,仍然需要更多的技术创新。
未来,单细胞蛋白质组学将继续发展,在技术创新和多组学整合的推动下,逐步克服这些挑战。未来的研究方向可能会集中在提高分析的灵敏度、提升样本通量以及开发更高效的数据分析方法上。随着这些问题的解决,单细胞蛋白质组学无疑将在精准医学、疾病早期诊断和治疗靶点的发现等领域发挥越来越重要的作用。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
